
Chapter 5: Devolatilization & Distillation (Decarboxylation & Distillation)
, by Avery Benitez, 57 min reading time
, by Avery Benitez, 57 min reading time

DEVOLATILIZATION

20L Double Jacket Glass Reactor By BVV
Devolatilization is often defined as the removal of volatile compounds within a substance. This is often necessary to further refine a botanical extract via distillation. Devolatilization is an evaporative process that drives volatiles from a botanical extract when heat is applied. Typically this is a separate process performed prior to distillation for the sake of efficiency.
Devolatilization as a process is used to remove volatile compounds from a botanical extract, convert compounds into a more distillable form, and ensure an extract is effectively purged of residual solvent prior to performing distillation. When a botanical extract is properly devolatilized, it results in a smoother, more efficient distillation of a botanical extract.
One of the more common forms of devolatilization of a botanical extract is decarboxylation. Decarboxylation is a chemical reaction that removes a carboxyl group from an organic compound releasing carbon dioxide. Typically decarboxylation is performed to remove loosely bonded carboxylic acids from the atomic chain of a botanical compound converting it into a more stable, easily distilled form. The most common use for decarboxylation is to convert acidic phytocannabinoids into their non-acidic active forms by evaporating the loosely bonded carboxyl group from the compound resulting in the release of CO2.

Begining of Devolatilizaiton
In this case, decarboxylation is necessary prior to distillation because the decarboxylation process occurs before the evaporation of the cannabinoid when heat is applied to the extract. This makes decarboxylation necessary to refine a cannabinoid-rich extract further into a distillate.
Both devolatilization and decarboxylation occur over time with the application of heat. The rate at which devolatilization is performed depends on how much heat is applied to the extract over time. While this process can be achieved quickly at high heat, this may evaporate some of the lower boiling point target compounds found within the matrix of a botanical extract. This is why most devolatilization processes call for moderate heat just below the lowest boiling point of the target compounds the operator is looking to distill. This results in the evaporation of all the lower boiling point compounds within the botanical extract, prepping it for an efficient distillation of the target compounds.
While this process can be performed within a short path distillation system prior to distillation, it is best to perform this process in a separate apparatus for processing efficiency and not allow volatiles to contaminate the vacuum pump of the short path distillation apparatus.

Partially Devolatilized Oil
Devolatilization of a botanical extract is typically performed inside a stainless steelor glass reactor by heating the extract just below the lowest boiling point of the target compounds within the botanical extract. While this can be accomplished at ambient pressures, devolatilization can be achieved more efficiently under vacuum with agitation. Devolatilization is typically performed by incrementally heating an extract under vacuum to the desired devolatilization temperature until no more volatiles evaporate from the solution. While devolatilization temperature is highly variable based on the target compounds of the distillation, they usually range between 120C to 150C / 248F to 302F. A good indication that a botanical extract is fully devolatilized is when extract consistency stabilizes, and boiling is no longer observed at the set devolatilization temperature. Once evaporation has ceased the devolatilization process is complete. The extract should be allowed to cool to 65C/149F before transferring the devolatilized extract to another vessel for further processing.
Devolitilization and Decarboxylation should always be performed prior to distillation, not only for the sake of processing efficiency but also for safety. When an extract that has not been decarboxylated or devolatilized is loaded into a distillation system and heat and vacuum are applied the extract tends to expand rapidly as CO2 releases causing a violent reaction. This can result in excessive boiling that can overflow and contaminate the system, the contamination of vacuum pump oil with volatiles, and the potential of pressure build-up resulting in an explosion of glass. For these reasons, it is always best to perform devolatilization in a separate apparatus. The devolatilization apparatuses can range from a Jacketed reactor, heated, stirring plate under a fume hood, or convection oven with appropriate venting.

Full Decarboxylated Crude Oil
DEVOLATILIZATION STANDARD OPERATING PROCEDURE
Purpose
The purpose of this procedure is to provide detailed instructions for the devolatilization of a botanical extract prior to distillation.
Scope
This procedure applies to all lab technicians tasked with botanical extract devolatilization.
Definitions/Acronyms
Personal Protection Equipment (PPE) Items worn to protect employees from exposure to hazardous materials and prevention of injury.
Safety Data Sheet (SDS) Provides useful information on chemicals, describing the hazards the chemical presents, and giving information on handling, storage, and emergency measures in case of an accident.
Devolatilization The removal of volatile compounds within a substance.
Decarboxylation A chemical reaction that removes a carboxyl group from a compound releasing CO2.
Safety
SDS Sheets: See extraction solvent SDS for detailed risks
PPE: The following should be worn by all lab personnel while winterizing botanical extracts:
Protective eyewear
Lab coat
Gloves
5. Hazard Identification
Preparation and Use:
A botanical extract will be heated to remove any volatiles remaining in the extract prior to distillation.
Concentration- >10% residual extraction solvent.
Quantity- Residual extraction solvent.
Frequency- No new solvent introduced.
Location- Jacketed reactor, headed stirring plate under a fume hood, or convection oven with appropriate venting.
Potential Hazards and Risks
See extraction solvent SDS for detailed risks.
The explosion from excessive internal pressure: the devolatilization could explode if the internal pressure produced by off-gassed volatiles becomes too great.
Burns: Do not touch heated surfaces flask during operation.
Broken glassware: Cracks or chips in glassware can result in glass rupturing causing chemical exposure and explosion hazard to the user. Always inspect glassware prior to use
6. Procedure
1. Transfer winterized and solvent recovered botanical extract into a devolatilization vessel ensuring extract volume does not exceed ⅔ the volume of the devolatilization vessel.
2. Apply incremental heat until a temperature just below the lowest boiling point of the target compounds you are preparing to distill is reached.
3. Continue to apply heat to the extract just below the temperature of your target compound's lowest boiling point until extract consistency stabilizes and boiling is no longer observed.
4. Allow your solution to cool to 65C/149F before transferring the extract to a new vessel.
DISTILLATION

5L Neocision Short Path Distillation Turnkey Kit By BVV
Distillation is a powerful purification technique that can drastically increase the purity of a botanical extract. Distillation can be simplified as the act of purifying a liquid by using selective evaporation and condensation. This purification process is implemented by heating the input material until target compounds are evaporated and then pulling the resulting vapor through a chilled condenser. It is condensed back into liquid form and collected. Distillation carries excellent utility in separating compounds in a mixture when there is a great enough difference between their boiling points and or their vapor pressure.
Vapor pressure or equilibrium vapor pressure is the pressure of a vapor in thermodynamic equilibrium within its condensed phase inside a closed container. When a liquid is confined to a closed container, an equilibrium exists between its liquid and its gaseous phase. This equilibrium exists regardless of the temperature inside the container or the liquid because some particles within the liquid, essentially at any temperature, will always have enough energy to escape the intrinsic cohesive forces and enter the gaseous state.
When distilling an impure mixture of compounds like that found within a botanical extract, the various constituents contribute to the overall vapor pressure of the liquid depending on the concentration of compounds within the solution and their respective boiling points. The vapor pressure of a solution is affected by temperature alone and is not a function of volume or surface area. As the temperature of a solution increases or decreases, so does its vapor pressure. During the distillation process, when the temperature and the vapor pressure of a compound have increased past the ambient pressure of the closed system it is within, a compound will transform from a condensed state to a gaseous state referred to as evaporation.

Evaporation Occurring
Equilibrium vapor pressure is an indication of a liquid's evaporation rate. It relates to the tendency of particles to escape from their condensed phase into the vapor phase, which can be observed as boiling. When a liquid is boiling, its vapor pressure is equal to or greater than the ambient pressure within the closed system containing it causing compounds to evaporate from their condensed state. The rate at which these compounds are evaporated is determined by its vapor pressure which is a function of the temperature exerted upon its liquified state and the ambient pressure of the closed system that it must overcome.
The evaporation of compounds happens at different rates depending on the vapor pressure of each constituent. Compounds with the highest vapor pressure tend to evaporate from the solution first as heat is applied during distillation. Compounds evaporate from the solution in order based on their atmospheric boiling point as heat is applied. Compounds with similar vapor pressures are commonly evaporated and condensed at the same temperature range making it challenging to isolate compounds in some instances completely.
Typically compounds within the same temperature range are evaporated, condensed, and collected into what is referred to as a fraction. These fractions are typically divided into three categories: the heads fraction containing the compounds that evaporate before the target compounds, the main body containing the desired target compounds, and the tails fraction containing the compounds that evaporate after the target compounds.
When it comes to evaporation, for a compound to undergo the phase change from a liquid to a gas, it needs to overcome the ambient pressure within the system. Think of ambient pressure as an omnipresent weight pushing down at all times. The greater the ambient pressure the more energy in the form of heat is needed for compounds to evaporate.

This can be observed when boiling water at different elevations or atmospheric pressures. Atmospheric pressure is the weight of the atmosphere exerted on the earth's surface. Typically measured with a barometer, at sea level where atmospheric pressure is greatest, barometric pressure is about 14.7 PSI under standard conditions. There is less atmospheric pressure at higher elevations, resulting in reduced resistance to evaporation. As a result, water is observed to boil at a lower temperature at higher elevations. It requires more energy in the form of heat in order to boil at sea level due to the increased atmospheric pressure.
By reducing the amount of ambient pressure exerted upon a compound we can drastically reduce the amount of resistance a compound must overcome to evaporate. While energy in the form of heat is necessary to trigger evaporation by reducing the pressure within the system by pulling it under a vacuum, the amount of energy in the form of heat necessary to induce evaporation is reduced. The most efficient way to reduce pressure within a closed system is to pull the system under vacuum with a vacuum pump.

21.2CFM Pro Series Corrosion Resistant Two Stage Vacuum Pump By BVV
Vacuum can be considered negative air pressure or pressure measuring below atmospheric pressure. Air pressure is defined as atmospheric pressure or the pressure within a container due to the compression of gases. When a system is pulled under a vacuum, the air pressure is evacuated until no air or gaseous volumes remain. This reduces the amount of pressure a compound needs to overcome to evaporate. When pressure is reduced, it, in turn, lowers the threshold of vapor pressure needed for a compound to evaporate, escaping its liquified state into a gaseous state.
This is beneficial when distilling botanical compounds because heating a botanical extract near the atmospheric Boiling points of its target compounds for prolonged periods can induce degradation in some instances. By applying vacuum to our distillation procedure, we can reduce resistance to evaporation and the amount of heat needed to evaporate the target compound from a botanical extract accelerating the process. Vacuum can be achieved to a varying degree depending on the strength of the vacuum pump and the design of the distillation apparatus with nonrestrictive flow being ideal.
Vacuum is measured with any set of force per area units. In the customary US system, vacuum is measured in inches of mercury, abbreviated Hg, the atomic symbol of mercury. This measurement is determined by using a mercury barometer by measuring the distance traveled by a pool of mercury up a tube when pulled by vacuum, with the maximum level of vacuum measured as 29.9 inches of mercury.
Within the context of distillation through a vacuum pump, the entire distillation apparatus is pulled under vacuum, reducing the ambient pressure of the system, which in turn reduces resistance to evaporation and the amount of vapor pressure needed to achieve the evaporation. The continuous operation of the vacuum pump through the distillation process also assists in pulling the ensuing vapors from the evaporator to the condenser portion of the distillation apparatus.

P10V6 Neocision Distillation Head By BVV
Condensation is the other half of the distillation process. Condensation is defined as the conversion of a vapor or gas to a liquid. Condensation is initiated by the formation of molecular clusters of vapor within a gaseous volume. Condensation occurs when a vapor is cooled or compressed past its saturation limit within a given volume.
Molecules within a vapor are typically far apart from one another. As more vapor collects within a given volume of air, it becomes saturated with vapor. This increase of density or closeness of vapor molecules increases the tendency of the molecular clustering of vapor, resulting in condensation.
The saturation limit of a gaseous volume is the maximum amount of vapor a given volume of air can contain. The saturation of a gaseous volume can be achieved via either evaporation or cooling. Evaporation achieves saturation of a gaseous volume by increasing the level of vapor in a given volume. As more vapor collects within a given volume, that volume reaches its maximum vapor capacity and is considered fully saturated with vapor.
Conversely, cooling can be utilized to reduce the saturation capacity of a gaseous volume and reach its saturation limit. Similar to the saturation of a solvent, a gaseous volume has a reduced saturation capacity at colder temperatures. Since a gaseous volume has less saturation capacity at lower temperatures cooling can be utilized to reach a gaseous volume's saturation limit and induce the molecular clustering of vapor.
When a given volume of air is saturated with vapor, the vapor density is at its maximum. With the vapor packed so densely, condensation is likely to be initiated by the formation of molecular vapor clusters of a compound within its gaseous volume or at the contact of vapor with a colder liquid or a solid surface. When a saturated gaseous volume is cooled past its saturation point, it tends to slow the movement of vapor and initiate the clustering of molecular vapor within the gaseous volume or on a surface at a lower temperature than itself, resulting in the vapor being condensed back into a liquid phase.

Valved Vacuum Adapter 2.0 By BVV
Based on the principles of psychrometry, the vapor’s condensation point present in a sample of air can be defined as the temperature at which the vapor changes into a liquid. The temperature value that allows this process is referred to as the dew point. The dew point of vapor is the temperature at which vapor will condense out of the gaseous phase into the liquid phase. In the context of distillation, vapor does not need to be chilled excessively to initiate condensation. Generally, any temperature at least 40C/104F degrees colder than the temperature at which the compound evaporated is sufficient In recondensing a vapor back into a liquid.
During the condensation of vapor into a liquid, as the vapor cools the gaseous volume reaches its maximum vapor capacity or condensation point and condenses into a liquid. In doing so, the latent heat is released by the vapor and transferred to the surrounding environment. In the context of distillation, the heat is absorbed by the transfer fluid of the condenser. A distillation condenser is typically a jacketed portion of the distillation apparatus circulated with chilled fluid creating a lower temperature portion of the unit where vapors can be condensed back into liquid form.
The condenser cools the ensuing distillation vapors down, slowing their movement, initiating the molecular clustering of vapor, and providing surface area for vapors to condense on. As vacuum pulls the distillation vapors through the condenser, the recirculated fluid absorbs the heat from the vapor, thus reducing its saturation capacity beyond the condensation point resulting in the condensation of vapor into a liquid. The resulting liquid formed from the distillation process is referred to as distillate. The condensed distillate flows down from the condenser into a collection apparatus. Once the desired target compounds have been distilled, the result should be a highly purified extract containing upwards of 90% of the target compound. This highly pure extract can be used to make a variety of different products or be used as input for further isolation of target compounds.
SHORT PATH DISTILLATION
5L Neocision Short Path Distillation Turnkey Kit By BVV
Short Path Distillation is a relatively low-cost distillation method that allows the user to collect separate fractions of distillate resulting in a versatile high purity botanical concentrate. Short path distillation utilizes a short distance to evaporate and recondense target compounds from a botanical extract. It is performed by applying heat and vacuum to a botanical extract to evaporate target compounds from the starting solution. The resulting vapor is then pulled by the vacuum pump's suction from the boiling flask, through the distillation head into the condenser where vapors are re-condensed back into a liquid form resulting in a highly concentrated distillate.
A typical short path distillation system consists of the following: a boiling flask and heated mantle where the input material is heated to the point of evaporation, a distillation head where vapor is refluxed and pulled through, a condenser chilled by a circulator where the ensuing vapor is recondensed back into liquid form, a receiving flask where the distilled compounds are collected, a cold trap to ensure all gaseous compounds are recondensed before reaching the final component a vacuum pump, which is typically monitored by a vacuum gauge.
While each component of the short path distillation system is essential to perform the process, the vacuum pump is most influential in the overall efficiency of the short path distillation process. The vacuum is used to pull the ensuing vapors through the short path distillation system and decrease ambient pressure within the short path distillation apparatus. When the system is pulled under vacuum, the pressure within the system is reduced, which lowers the amount of ambient pressure a compound must overcome in order to evaporate. By pulling the system under vacuum, resistance to evaporation is drastically reduced, increasing the efficiency of the evaporative process.

21CFM Dual-Stage High Capacity Vacuum Pump By Edwards
The level of vacuum depth a short path distillation apparatus can achieve is attributed to the performance and strength of the vacuum pump, along with the proper assembly & configuration of the short path distillation system. The configuration of the short path distillation system should provide minimal restriction to vapor flow, high CFM from the vacuum pump, and extremely low cold trap temperatures. Beyond configuration, if the vacuum pump oil should be fresh and adequately warmed up and the system has been properly assembled with no vacuum leaks, Short path distillation setups can achieve extremely low vacuum levels during distillation.
The performance of the vacuum pump during distillation can also be attributed to the effectiveness of the cold trap. Before the vacuum pump, the cold trap is the final component utilized to recondense any vapors that were not condensed by the initial condenser. A cold trap is typically filled with a dry ice/ethanol slurry or a chilled immersion probe to create an extremely low-temperature zone that is meant to recondense any vapors that make it past the initial condenser. This step ensures no vapors make their way into the vacuum pump, potentially contaminating the vacuum pump oil and reducing the pump's overall level of performance. Ideally, the cold trap should be chilled as cold as possible, with an ideal temperature being around -80C/-112F

Ribbed Glass Cold Trap By BVV
While the performance of the cold trap, the seal of the short path distillation apparatus, and the quality and temperature of the vacuum pump oil play a role in the achievable vacuum depth, these factors are limited by the strength of the short path distillation vacuum pump. Typically a rotary vane vacuum pump pulls the short path distillation apparatus under vacuum due to its ability to achieve deep vacuum levels. To ensure a rotary vane vacuum pump is operating at peak efficiency and quality distillation is achieved it is best to utilize fresh vacuum pump oil for every distillation run as contaminated vacuum pump oil can prevent a vacuum pump from reaching the deep vacuum levels that are ideal for short path distillation.

Premium Vacuum Pump Oil By BVV
The vacuum pump works with the heated mantle to perform the evaporative process during distillation. While vacuum drastically increases the efficiency of the evaporative process, energy in the form of heat is necessary in order to trigger the evaporative process of distillation. During short path distillation, evaporative energy is produced by the heated mantle. The heated mantle is an apparatus designed to house and efficiently apply heat to the boiling flask. The heated mantle can be controlled to deliver precise heat to the boiling flask by monitoring the internal temperature of the solution through the use of a thermal probe that is inserted into the boiling flask and submerged into the solution. The thermal probe allows for precise heat application to the starting solution allowing for controlled evaporation of target compounds.

20L Digital Heating Mantel By BVV
The heated mantle commonly performs both the essential application of heat to the distillation process and provides agitation of the solution through magnetic stirring. Heated mantle typically comes equipped with a magnetic stirring function that allows for control over the heating and agitation of the solution within the boiling flask. This is accomplished through the use of a magnetic stir bar that is submerged into the solution during the loading of the botanical extract into the boiling flask. The heated mantle controls the rate at which the magnetic stir bar rotates, which results in how much the solution is agitated. Agitation of the solution increases heat transfer, helps vapor bubble formations escape from the solution more easily and attributes to vapor momentum to some extent. As a compound is evaporated from a solution, bubble formations deeper within the liquid, typically require more energy to overcome the higher fluid pressure. When agitation is applied to the solution, the rate at which vapor bubbles escape the solution is accelerated, and the rate of boiling is increased.
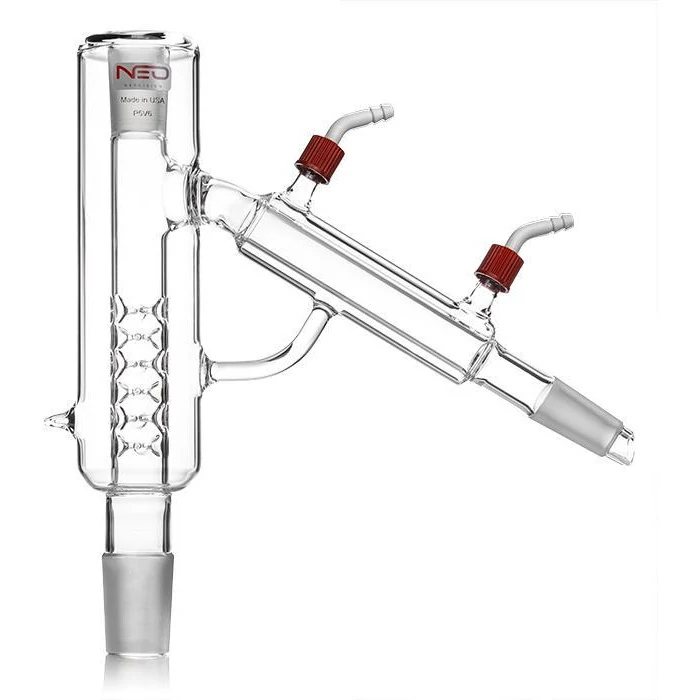
P5V6 Neocision Distillation Head By BVV
As the solution boils, the ensuing vapors are pulled via the suction of the vacuum pump out of the boiling flask into the distillation head. The distillation head typically contains a complex pathway of glass vigreuxs that act as a "theoretical plate" or section of friction where heavier compounds with less evaporative energy can attach to, recondense, and fall back into the boiling flask, which is referred to as reflux. The refluxing of heavier compounds with less evaporative energy results in a purer vapor stream of lighter compounds, with great enough evaporative energy to pass through the distillation head into the condenser.

Traceable Kangaroo Digital Thermometer By Thermo Scientific
While the primary function of the distillation head is to accomplish the separation of heavier and lighter vapors to create a more pure vapor stream and resulting distillate flow, the distillation head also serves as a point where vapor temperature can be measured through the use of a thermal probe. Distillation heads commonly contain a port where a thermal probe can be inserted, allowing for the invaluable measurement of the distillation head vapor temperature. Distillation head vapor temperature Is an incredibly valuable tool in measuring the evaporative process. Observation of a drop in head vapor temperature or an observed decreased boiling rate is a great indicator that evaporation at the set temperature has decreased and mantle temperature can be increased to continue the evaporative process.

Stirmantle By Glas-Col
During the evaporative process, as mantle temperature is increased, it subsequently increases head vapor temperature. Over time as the solution boils at the set mantle temperature, the compounds with corresponding vapor pressure evaporate from the solution resulting in a reduction of vapor being created over time, which can be observed visually as a decreased rate of boiling and an overall drop in head vapor temperature. When a 0.5C decrease in head vapor temperature is observed, it can be used as an indicator that the rate of evaporation of compounds in that temperature range has decreased, and mantle temperature can be increased to continue the evaporative process and ensure a timely distillation. By incrementally increasing mantle temperature based on dips in distillation head vapor temperature, the operator can achieve the most efficient distillation possible.
Conversely, an experienced operator can establish timed intervals to increase temperatures or increase the mantle temperature as it warms by defined increments. Short path distillation can be performed successfully with various methodologies that are commonly centered around increasing mantle temperature incrementally to separate compounds via selective evaporation, condensation, and collection.

P10V6 Neocision Distillation Head - USA Made By BVV
During distillation, the ensuing vapors that make their way through the distillation head arrive at the condenser portion of the short path distillation apparatus. This jacketed glass portion of the short path distillation system allows for heated or chilled thermal fluid to be recirculated through the jacket utilizing a heated circulator. This lower temperature portion of the short path distillation apparatus is utilized to recondense the vapors that make it through the distillation head so the resulting distillate can be collected into the receiving flask.
Contrary to popular belief, the condenser does not need to be extremely cold to effectively condense vapors; typically a 20C/68F colder condenser temperature than the vapor temperature is sufficient in condensing distillate vapors. While cold condenser temperatures can be ideal when distilling volatile compounds like terpenes, too cold of a condenser can increase the viscosity of the distillate stream, which can result in the clogging of the condenser. Conversely, too hot of a condenser temperature can result in vapor flowing through the condenser without being condensed.

5 Liter Neocision Heated Circulator By BVV
During distillation, the condenser circulator temperature can be set using a variety of different strategies, including leaving the condenser at the same temperature through the entirety of the distillation procedure or incrementally increasing the circulator temperature to maintain a 20-degree colder temperature than the vapor temperature up to the heated circulator’s limit and lines while not exceeding a heated recirculation temperature of 130C/266F. While both methods can produce excellent results, the latter is recommended for novice operators with an ideal heated circulator temperature range between 70-90C/158-194F being sufficient for condensing vapors through the entirety of the distillation.
Depending on the short path distillation apparatus after the condenser, there can be a variety of different collection apparatuses utilized for capturing different fractions from the distillation process ranging from a single collection vessel with an isolation valve, a multi-position cow, a swing arm adaptor, or a discharge pump. While swing arm adaptors and multi-position cows are great for collecting multiple fractions without breaking vacuum, isolation valves and discharge pumps allow for the capturing an infinite amount of fractions during distillation with a discharge pump allowing for ultimate efficiency for high volume distillers. Regardless of the collection apparatus, the goal of all these components is to allow for the collection and separation of distillate fractions.

Outlet Pump By Beaker & Wrench
Short path distillation is commonly used as a form of fractional distillation by incrementally increasing the heat applied by the mantle to distill and separate compounds based on their difference in boiling points and or vapor pressures. The boiling point of a substance is the temperature at which a pure substance evaporates, observed as boiling under normal atmospheric conditions. When dealing with a mixture of many compounds like that of a botanical extract, each compound contributes a fraction of the total vapor pressure based on their respective boiling points, known as partial vapor pressure.

Evaporation inside a boiling flask
Typically the lower a compound's boiling point, the higher its vapor pressure. Once the vapor pressure of a compound exceeds the ambient pressure of the closed system containing it, that compound will evaporate from the solution observed as boiling. As the temperature of the solution is increased, vapor pressure is also increased, resulting in compounds evaporating from the solution in order based on their respective boiling points, starting with the most volatile compound with the lowest boiling points.
During distillation, compounds with similar boiling points tend to evaporate within the same temperature range, making it difficult to completely isolate compounds. While complete isolation of a certain compound is typically easier to achieve through crystallization or chromatography. Fractional distillation can be utilized to efficiently separate compounds with different boiling point ranges and collect highly pure fractions of compounds that evaporate within the same temperature range.

2L Short Path Distillation Kit By BVV
Typically, these fractions are collected into three categories: the heads fraction, the main body fraction, and the tails fraction. The heads fraction contains the compounds first evaporated from the solution, consisting of the compounds with a lower boiling point than that of the target compounds. As the heated mantle temperature increases and the target compounds start to evaporate from the solution, the main body fraction starts to distill. At this point of the distillation, the collection or receiving flask is changed in order to separate the lower boiling point compounds within the heads fraction from the target compounds of the distillation to collect a higher purity fraction of the target compounds referred to as the main body.

Fraction Finder By Arometrix
During distillation, it can be challenging to pinpoint the appropriate time to change flasks between the heads and main body fraction. While a UV Detector can be utilized to pinpoint which compounds are being distilled without this type of device, it is commonly left to the observation of distillate flow. Typically If distillate viscosity is increased or If analytical results show that the starting input predominantly consists of the target compound and an increased flow of distillate is observed, both can be a good indicator that the target compounds are starting to distill and the collection flask should be changed to isolate the main body from the heads fraction.

Heads Fraction
To ensure the main body fraction is of the highest purity, it is common to allow some of the main body to collect into the heads fraction before changing flasks. This ensures that the lighter volatile compounds are not collected into the main body fraction. Once the main body is assumed to be distilling before swapping the flask, it's best to allow evaporation at that temperature range to diminish, observed by a significant drop in head vapor temperature and a visual reduction of boiling to ensure the lower boiling point compounds have distilled before swapping collection flasks and increasing mantle temperature to evaporate the target compounds of the main body fraction at a higher temperature range.
As the distillation process is resumed and the evaporation of the main body is achieved. Once satisfied with the collection of the main body, distillation can be stopped leaving the tails fraction remaining in the resinous remains of the starting material, or the main body flask can be changed, and the tails fraction can be evaporated from the solution into a separate collection vessel. While the tails fraction typically contains some of the target compounds as distillation temperatures increase, color pigments tend to darken the distillate flow that most distillers prefer to isolate from the main body fraction in order to keep the main body of the highest purity.
Fractional distillation is a process refined by the operator over time. While the ideal separation of distillate fractions can be hard to pinpoint, the process can be optimized through analytical testing, trial, and error. At the same time, the separation of fractions is dependent on the target compounds of the distillation. As a general rule of thumb, an increased rate of distillate flow and viscosity are good indications the distillation is entering the main body fraction, and a visual darkening of the distillate flow can indicate the tails fraction is starting to distill.
Before the process of short path distillation can be initiated, the apparatus must first be appropriately staged and properly assembled. A short path distillation system is meant for operation within a fume hood or within a well-ventilated area. When staging a short path distillation apparatus, assure the system is assembled in an area where the vacuum pump exhaust can be vented outside the facility.
When a suitable area has been identified, short path distillation assembly starts with positioning the heated mantle and support stands. For greater ease of use, the heated mantle of a short path distillation apparatus is commonly elevated to allow for more clearance when changing the collection flask to separate fractions. After staging the mantle, preassemble all lab stands, boss heads, three-finger clamps, chain clamps, scissor jacks, and make sure to have keck clips and vacuum grease within arms reach to aid in the assembly of glassware.

Laboratory Scissor Jack By BVV
Proper assembly of the short path distillation apparatus is essential for producing the deep vacuum levels that are ideal for short path distillation. To assure the deepest possible vacuum is achieved, it is important that each joint of the short path distillation system is appropriately assembled with vacuum grease to create a complete seal between joints. When choosing a vacuum grease for application, ensure the grease is rated for the temperatures of the distillation. For temperatures above 204C/399F Apizon High-Temperature Vacuum Grease is highly recommended.
Vacuum grease is used to create a perfect seal between the separate components of the short path distillation apparatus. To properly grease each joint, place a small amount of vacuum grease on the upper half of the male joint and insert it into the female joint. Once in place, rotate the male joint until an even coating of vacuum grease can be seen across the entire circumference of the upper half of the male joint. When a complete seal of vacuum grease has been made, secure the joint with an appropriately sized keck clip and support the component with the appropriate lab stand support or scissor jack. Keep in mind that vacuum grease should only be applied to the upper half of the male joint to ensure vacuum grease does not come in contact with the lower half of the joint, potentially contaminating the inside of the system.

High-Temperature Vacuum Grease By Apiezon
During the assembly process, it is important to ensure all glass joints are appropriately greased with vacuum grease, creating a complete seal, and each component is fully supported by the appropriate lab stands or scissor jacks. To start the process of assembly, first place the boiling flask inside the heating mantle, and install the mantle temperature probe and connect the distillation head to the boiling flask. Once the distillation head has been properly attached, install the distillation head temperature probe and proceeds to attach the condenser if it isn't already integrated into the distillation head. After the condenser has been properly installed and supported, proceed to assemble the remainder of the short path distillation glassware prior to connecting the condenser to the heated circulator to ensure joints can be manipulated into place.
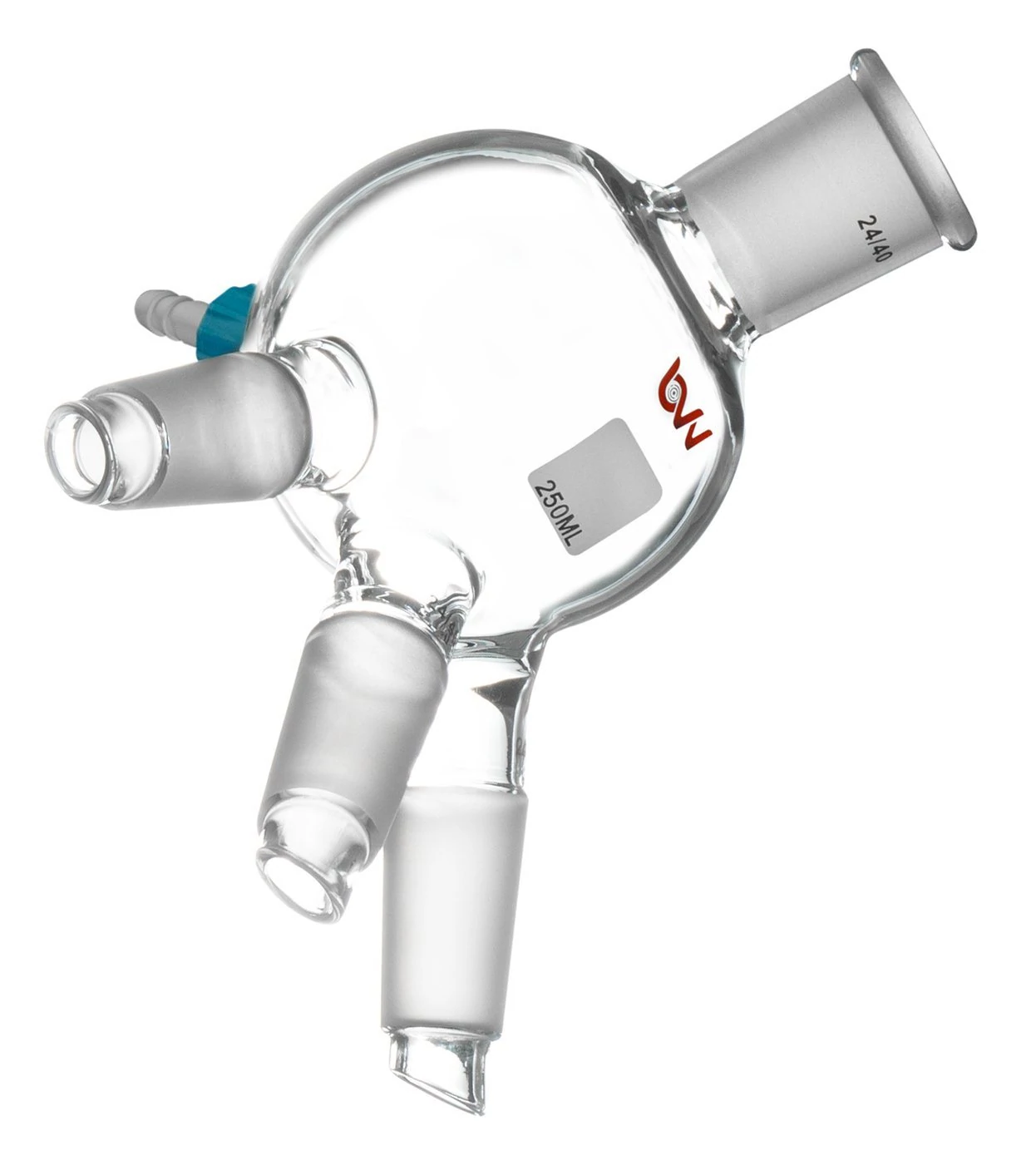
Triple neck Cow Receiving Flask By BVV
Continue assembly by connecting the condenser to the collection apparatus and installing the receiving flasks making sure each component is connected properly and supported using the appropriate lab stand or scissor jack supports. After assembly of the collection apparatus, proceed to connect the cold trap ensuring all connections are appropriately greased, and the cold trap is properly supported using a chain clamp. Once the cold trap has been properly assembled, proceed to connect the vacuum gauge and vacuum pump using the appropriate vacuum-rated lines and plumb the vacuum pump exhaust outside the facility.
Now that the short path distillation apparatus has been completely assembled before proceeding to vacuum test the system, prior to doing so, double-check to ensure all glassware is appropriately supported by the lab stands and all components are properly connected. Proceed to vacuum test the system by first ensuring the vacuum pump is appropriately filled with fresh vacuum pump oil and closing the vacuum valve to the system before turning on the vacuum pump in open gas ballast mode and allowing it to warm up for 30 minutes to an hour. Once the vacuum pump oil has been appropriately warmed up, proceed to open the vacuum valve allowing the entire system to be pulled under vacuum.

Bullseye Precision Vacuum Gauge By DigiVac
At this point, monitor the vacuum gauge. Vacuum gauges provide invaluable monitoring of vacuum pressure during distillation, typically vacuum gauges are utilized after the cold trap, with an additional vacuum gauge before the cold trap providing data on the effectiveness of the cold trap. While handheld gauges provide a cost-effective monitoring solution, precision vacuum gauges are ideal for professional distillers allowing for precise monitoring of vacuum pressure.
During the initial vacuum pull, if deep vacuum levels are not attained, proceed to identify where the vacuum may be leaking by spraying each glass joint with soapy water, turning off the vacuum pump, and bleeding pressure before disassembling and reassembling connections that are not maintaining a seal. Proceed to vacuum test and disassemble and reassemble the system as needed until there are no vacuum leaks and vacuum levels can be maintained.
Once assembly is complete, and the short path distillation apparatus has been proven to maintain vacuum levels, it's best to test the achievable amount of vacuum depth by allowing the system to be pulled under vacuum in closed ballast mode. Given the system is properly assembled, In closed ballast mode, a rotary vane vacuum pump should be able to achieve vacuum depths below 25 microns with even lower depths being achievable prior to loading the system with the botanical extract.

2L Premium Short Path Distillation Kit By BVV
After the vacuum testing is complete, the last task is to connect the heated circulator to the condenser port and fill the heated circulator with the appropriate heat transfer fluid. If operating the heated circulator in temperature ranges below 90C/194F, a distilled water or water-glycol mixture is sufficient. If operating a heated circulator above 90C/194F, a high-temperature heat transfer fluid like silicone oil is recommended. When connecting the heated circulator to the condenser, ensure the heat transfer fluid inlet is flowing from the top of the condenser to the bottom, and all clamps are tightened appropriately.
The short path distillation system is now ready for operation. To prepare for short path distillation, start by bringing the system to temperature and allowing the vacuum pump to warm up in open ballast mode. Again ensure the vacuum pump oil is filled to the appropriate operating level and the vacuum valve to the system is closed, before proceeding to turn on the vacuum pump in open ballast mode and allowing it to warm up for 30 minutes to an hour before operation while the rest of the system is prepared.

21.2CFM Corrosion Resistant Two Stage Vacuum Pump By BVV
While the vacuum pump is warming up in open ballast mode, proceed to bring the cold trap to operating temperature by turning on the cold trap immersion probe chiller setting it to its lowest setting, or by preparing the cold trap with dry ice and ethanol slurry by filling the cold trap 1/2 way with ethanol and slowly adding dry ice to avoid over the bubbling of the solution.
As the cold trap reaches operating temperature, proceed to bring the condenser's heated circulator to operating temperature by first ensuring that the circulator bath is filled to the appropriate fill level and the fluid circulation is connected to flow into the top of the condenser and out of the bottom of the condenser. Proceed to ensure all clamps are tightened appropriately before turning on the circulator and setting it to the desired condenser temperature. For the start of the run, a temperature range between 40-50C/104F-122F is effective at condensing volatile compounds. Once volatiles have diminished a condenser temperature of 70-90C/158-194F can be used and maintained for the entirety of the run, or condenser temperature can be increased from this point to maintain a 20C/68F degree colder difference between the vapor temperature and the condenser temperature.

Digital Heating Mantle Probe By BVV
As the vacuum pump warms up and the system reaches the desired operating temperature, proceed to load the botanical extract into the boiling flask by removing the mantle thermal probe, cleaning the joint of any vacuum grease, and attaching a glass funnel. Proceed to fill the boiling flask with winterized and devolatilized botanical extract. The boiling flask should be filled to a minimum of 25% of the capacity of the boiling flask, ensuring that there is enough solution for the thermal probe to read, and no more than 50% of the total volume of the boiling flask to ensure the there is enough headspace for the solution to boil. Keep in mind winterized and devolatilized botanical extract is ideal for short path distillation as unwinterized crude oil will make cleaning the boiling flask difficult, and non-devolitized crude may impede its ability to achieve ideal vacuum levels.

Fabric Insulating Top By Glas-Col
Once the botanical extract has been loaded into the boiling flask, add the magnetic stir bar and reinstall the mantle temperature probe into the boiling flask ensuring the probe reaches 1/4" from the bottom of the boiling flask without touching the bottom of the flask or interfering with the rotation of the magnetic stir bar. The system is now sealed and can be vacuum tested once again to ensure a deep vacuum is reached before applying heat to the system. Proceed to switch the vacuum pump into closed gas ballast mode and slowly open the vacuum valve to pull the system under vacuum. Ideally, a vacuum range below 50 microns is ideal, with the deeper the vacuum depth the better. At this time before heat is applied to the mantle it is highly recommended to apply insulation to the boiling flask utilizing an appropriate boiling flask jacket or insulation rope to ensure boiling flask temperatures are maintained during distillation.

2L Premium Short Path Distillation Kit By BVV
During this time, an ideal vacuum pressure before filling and then poor vacuum performance after filling will likely cause an incomplete devolatilization. If this is the case, allow the oil to finish devolatilization and vacuum performance to improve before proceeding with distillation. Start by turning on the heated mantle setting the mantle temperature to 50C/122F and the magnetic stir RPM on a low until the viscosity of the botanical extract solution has decreased enough for mixing to occur. During the distillation process, both heated mantle temperature and magnetic stir bar RPM can increase the rate of evaporation. Keep in mind the rate of evaporation should never exceed the capability of the condenser or cold trap. For this reason a steady increase of heat should be used and stir bar RPM should be optimized to create ideal turbulence of the liquid and maintained during the entirety of the run.

28mm L-Style Magnetic PTFE Stirrer By BVV
Once the heated mantle reaches the set temperature of 50C/122F, proceed to increase mantle temperature in 20C/68F increments as the mantle operating temperature reaches the set temperature point until the extract is observed to boil. Once boiling is observed, mantle temperature can now be increased based on an observed drop in distillation head vapor temperature or using a decreased rate of boiling or reflux as indicators to increase mantle temperature. For the latter, once a 1-degree drop in distillation head vapor temperature is observed, proceed to increase mantle temperature in 5C/41F degree increments until the time comes to swap collections flasks and collect a different fraction.

2L Digital heating and Stirring Mantle By BVV
It is highly recommended to increase slowly and steadily as to prevent bumping over or over-reacting inside the boiling flask. Short path distillation can be slow to start, and the operator should maintain presence and monitor the system closely as temperatures are increasing. Keep in mind that during this time, as distillation head vapor temperature increases, condenser temperature can also be increased to maintain a 20C colder condenser than the distillation head vapor temperature, or it can be kept at the same temperature for the entirety of the distillation procedure, typically between a range of 70-90C/158-194F.
As distillation continues, when distillate viscosity is increased, or an increased flow of distillate is observed, both can be a good indicator that the target compounds are starting to distill, and the collection flask should be changed to isolate the main body from the heads fraction.
To ensure the main body fraction is of the highest purity, it is common to allow some of the main body to collect into the heads fraction before swapping flasks; this ensures that the lighter volatile compounds are not collected into the main body fraction. Once the main body is assumed to be distilling prior to swapping the flask, it's best to allow evaporation at that temperature range to diminish, observed by a significant drop in head vapor temperature and a visual reduction of boiling to ensure the lower boiling point compounds have distilled prior to swapping collection flasks and increasing mantle temperature to evaporate the target compounds of the main body fraction at a higher temperature range.

Valved Vacuum Adapter 2.0 By BVV
When it comes time to swap flasks and separate fractions, this process can differ depending on the collection apparatus. When using an isolation valve, start by turning the isolation valve clockwise to isolate the receiving flask, then open the bleeder valve to break the vacuum on the receiving flask before proceeding to change flasks. Once the flasks have been changed first seal the bleeder valve before slowly turning the isolation valve counterclockwise to connect the collection flask back to the system and proceed to evaporate and collect the main body fraction.
As the distillation process is resumed and the evaporation of the main body is achieved. Continue to ramp mantle temperature in 5C increments as a 1-degree drop in head vapor temperature is observed. Depending on personal preference, once satisfied with the collection of the main body, distillation can be stopped leaving the tails fraction remaining in the resinous remains of the starting material or the main body flask can be changed yet again to capture the tails fraction of the distillation. Typically a visual darkening of condensate is used as an indication to swap flask yet again to collect the tails fraction in a separate fraction. While the tails fraction can contain some desired compounds, these are typically separated from the main body to achieve a main body of the highest purity and preserve a lighter colored main body.
Once satisfied with the collection of either the main body of the tails fraction, proceed to end the distillation run by first reducing the mantle temperature set point to 100C/212F. As the mantle temperature falls below 150C/302F, proceed to close the vacuum valve, turn the vacuum pump off, and slowly release vacuum from the system allowing system pressure to equalize. The collection flask can now be disassembled and transferred to a storage vessel. Before transferring the distillate, ensure the vacuum grease is cleaned from the collection flask joints before transferring the distillate to another vessel. Now that the distillate has been collected it can be redistilled which is referred to as a second pass to further refine the main body fraction resulting in an even purer collection of target compounds or the first pass distillate can be used for further refinement via isolation.

SHORT-PATH DISTILLATION STANDARD OPERATING PROCEDURE
Purpose
The purpose of this procedure is to provide detailed instructions for short path distillation of a botanical extract.
Scope
This procedure applies to all lab technicians tasked with botanical extract distillation.
Definitions/Acronyms
Personal Protection Equipment (PPE): Items worn to protect employees from exposure to hazardous materials and prevention of injury.
Safety Data Sheet (SDS): Provides useful information on chemicals, describing the hazards the chemical presents, and giving information on handling, storage, and emergency measures in case of an accident.
Distillation: is The action of purifying a liquid by a process of heating and cooling.
Decarboxylation: is a chemical reaction that removes a carboxyl group from an organic compound resulting in the release of carbon dioxide and the conversion of a compound into a more distillable form.
Vacuum: a vacuum is created by removing air from a container using a vacuum pump evacuating the space entirely of matter.
Micron/mTorr: are interchangeable units of measure for vacuum depth represented as one-millionth of a meter of mercury displacement under vacuum.
Safety
SDS Sheets: No additional chemicals are used in this process.
PPE: The following should be worn by all lab personnel while performing botanical extract distillation:
Protective eyewear
Lab coat
Gloves
5. Hazard Identification
Preparation and Use:
Concentration- Winterized and decarboxylated botanical extract.
Quantity- >60% boiling flask volume.
Frequency- An initial volume of the botanical extract is used, no more are added.
Location- Distillation occurs inside a short path distillation still under vacuum within a well-ventilated area.
Potential Hazards and Risks
The explosion from excessive internal pressure: the evaporator flask and/or the condenser could explode if the internal pressure produced by evaporation becomes too great.
Burns: Do not touch the heated mantle or evaporating flask during operation.
Broken glassware: Cracks or chips in glassware can result in glass rupturing causing chemical exposure and explosion hazard to the user. Always inspect glassware prior to use.
6. Preparation
Assembly
Stage the short path distillation apparatus in a well-ventilated area, where the vacuum pump exhaust can be vented outside the facility.
Assemble the short path distillation system, ensuring each joining is properly greased by applying a small amount of vacuum grease on the upper half of the male joint and inserting it into the female joint. Once in place, rotate the male joint 360 degrees until an even coating of vacuum grease can be seen across the entire circumference of the upper half of the male joint. (For distillations at temperatures above 204C/399F, use Apizon High-Temperature Vacuum Grease.)
When a complete seal of vacuum grease has been made, secure the joint with an appropriately sized keck clip and support the component with the appropriate lab stand support or scissor jack.
Ensure all process connections are appropriately sealed, and all glassware is appropriately supported by the appropriate lab stands and scissor jacks and proceed to vacuum test the system.
Ensure the vacuum pump is appropriately filled with fresh vacuum pump oil and close the vacuum valve to the system before turning on the vacuum pump in open gas ballast mode and allowing it to warm up for 30 minutes to an hour.
Once the vacuum pump oil has been appropriately warmed up, proceed to open the vacuum valve allowing the entire system to be pulled under vacuum.
At this point, monitor the vacuum gauge, if deep vacuum levels are not attained, proceed to identify the vacuum leak by spraying each glass joint with soapy water, turning off the vacuum pump, and bleeding pressure before disassembling and reassembling connections that are not maintaining a seal.
Proceed to vacuum test and disassemble and reassemble the system as needed until there are no vacuum leaks and vacuum levels can be maintained.
After the vacuum testing is complete, proceed to connect the heated circulator to the condenser port and fill the heated circulator with the appropriate heat transfer fluid. If operating the heated circulator in temperature ranges below 90C/194F, distilled water or a water-glycol mixture is sufficient. If operating a heated circulator above 90C/194F, a high-temperature heat transfer fluid like silicone oil is recommended. When connecting the heated circulator to the condenser, ensure the heat transfer fluid inlet is flowing from the top of the condenser to the bottom and all clamps are tightened appropriately.
Preparation
To prepare for short path distillation, start by allowing the vacuum pump to warm up in open ballast mode and bringing the system components to operating temperature.
Ensure the vacuum pump oil is filled to the appropriate operating level and the vacuum valve to the system is closed. Proceed to turn on the vacuum pump in open ballast mode and allow it to warm up for 30 minutes to an hour before distillation while the rest of the system is prepared.
While the vacuum pump is warming up in open ballast mode, proceed to bring the cold trap to operating temperature by turning on the cold trap immersion probe chiller setting it to its lowest setting, or by preparing the cold trap with a dry ice/ethanol slurry by filling the cold trap 1/2 way with ethanol and slowly adding dry ice to avoid over bubbling of the solution.
As the cold trap reaches operating temperature, proceed to bring the condenser's heated circulator to operating temperature by first ensuring that the circulator bath is filled to the appropriate fill level and the fluid circulation is connected to flow into the top of the condenser and out of the bottom of the condenser.
Proceed to ensure all clamps are tightened appropriately before turning on the system and setting it to the desired condenser temperature. For the start of the run, a temperature of 40C/104F can be used, and this incrementally increased, as vapor temperature rises to maintain a 20C degree difference between vapor temperature and condenser temperature, or the condenser can be set to a temperature range of 70-90C/158-194F for the entirety of the run.
As the vacuum pump warms up and the system reaches the desired operating temperature, proceed to load the botanical extract into the boiling flask by removing the mantle thermal probe, cleaning the joint of any vacuum grease, and attaching a glass funnel. Proceed to fill the boiling flask with winterized and devolitlized botanical extract. The boiling flask should be filled to a minimum of 25% of the capacity of the boiling flask, ensuring the there is enough solution for the thermal probe to read, and no more than 50% of the total volume of the boiling flask to ensure the there is enough headspace for the solution to boil. Keep in mind winterized and devolitilized botanical extract is ideal for short path distillation as unwinterized crude oil will make cleaning the boiling flask a pain, and non-devolitized crude may impede its ability to reach low vacuum levels.
Once the botanical extract has been loaded into the boiling flask, add the magnetic stir bar and reinstall the mantle temperature probe into the boiling flask ensuring the probe reaches 1/4" from the bottom of the boiling flask without touching the bottom of the flask or interfering with the rotation of the magnetic stir bar. The system is now sealed and can be vacuum tested once again to ensure a deep vacuum is reached before applying heat to the system.
Once the vacuum pump has operated in closed ballast mode for a minimum of 30 minutes, proceed to switch the vacuum pump into closed gas ballast mode and slowly open the vacuum valve to pull the system under full vacuum and start the short path distillation procedure.
6. Procedure
Turn on the heated mantle and apply heat to begin the process of distillation. Start by setting the mantle temperature to 50C/122F and the magnetic stir RPM on a low until the viscosity of the botanical extract solution has decreased enough for mixing to occur. During the distillation process, both heated mantle temperature and magnetic stir bar RPM can be used to increase the rate of evaporation. Keep in mind the rate of evaporation should never exceed the capability of the condenser or cold trap. For this reason, a steady increase of heat should be utilized, and stir bar RPM should be optimized to create ideal turbulence of the liquid and maintained during the entirety of the run.
Once the heated mantle reaches the set temperature of 50C/122F, proceed to increase mantle temperature in 20C/68F increments as the mantle operating temperature reaches the set temperature point until the extract is observed to boil.
Once boiling is observed, mantle temperature can now be increased based on an observed drop in distillation head vapor temperature or using a decreased rate of boiling or reflux as indicators to increase mantle temperature. For the latter as a 1C or a 5F degree drop in distillation head vapor temperature is observed, proceed to increase mantle temperature in 5C/41F degree increments until the time comes to swap collections flasks and collect a different fraction.
During this time, as distillation head vapor temperature increases, condenser temperature can also be increased to maintain a 20C colder condenser than the distillation head vapor temperature, or it can be kept at the same temperature for the entirety of the distillation procedure, typically between a range of 70-90C/158-194F.
When distillate viscosity is increased, or an increased flow of distillate is observed, both can be a good indicator that the target compounds are starting to distill, and the collection flask should be changed to isolate the main body from the heads fraction.
To ensure the main body fraction is of the highest purity it allows some of the main body to collect into the heads fraction before swapping flasks; this ensures that the lighter volatile compounds are not collected into the main body fraction.
Once the main body is assumed to be distilling prior to swapping the flask, allow evaporation at that temperature range to diminish, observed by a significant drop in head vapor temperature and a visual reduction of boiling to ensure the lower boiling point compounds have distilled prior to swapping collection flasks and increasing mantle temperature to evaporate the target compounds of the main body fraction at a higher temperature range.
When it comes time to swap flasks and separate fractions, this process can differ depending on the collection apparatus. When using an isolation valve, start by turning the isolation valve clockwise to isolate the receiving flask. then open the bleeder valve to break the vacuum on the receiving flask before proceeding to change flasks. Once the flasks have been changed first seal the bleeder valve before slowly turning the isolation valve counterclockwise to connect the collection flask back to the system and proceed to evaporate and collect the main body fraction.
As the distillation process is resumed and the evaporation of the main body is achieved, continue to ramp mantle temperature in 5C increments as a 1-degree drop in head vapor temperature is observed. Depending on personal preference, once satisfied with the collection of the main body, distillation can be stopped leaving the tails fraction remaining in the resinous remains of the starting material, or the main body flask can be changed yet again to capture the tails fraction of the distillation. Typically, a visible darkening of condensate is used as an indication to swap flasks and collect the tails fraction in a separate fraction. While the tails fraction can contain some desired compounds, these are typically separated from the main body to achieve a main body of the highest purity and preserve a lighter-colored main body. Keep in mind that distillation should not be continued past a mantle temperature of 220C/428F.
Once satisfied with the collection of either the main body or tails fraction, proceed to end the distillation run by first reducing the mantle temperature set point to 100C/212F.
As the mantle temperature falls below 150C/302F, proceed to close the vacuum valve, turn the vacuum pump off, and slowly release the vacuum from the system allowing system pressure to equalize.
The collection flask can now be disassembled and transferred to a storage vessel. Before transferring the distillate, ensure the vacuum grease is cleaned from the collection flask joints before transferring the distillate to another vessel.
The refined distillate can now be redistilled to refine the main body fraction resulting in an even purer collection of target compounds or be used for further isolation of target compounds via isolation.



